編者按:本文在戈登貝爾獎得主名單公佈後,於 11 月 17 日更新。
戈登貝爾高效能運算 COVID-19 研究特別獎的得主教導大型語言模型 (LLM) 一種新語言 (基因序列),因此能取得基因組、流行病學和蛋白質工程的相關見解。
這項突破性的技術於 10 月份發表,是由來自美國阿貢國家實驗室、NVIDIA、芝加哥大學等單位的二十多位學術與商業研究人員合作完成。
研究團隊訓練 LLM 追蹤基因變異,並預測可能在 COVID-19 之後出現的 SARS-CoV-2 病毒變異株。雖然目前大多數應用於生物學的 LLM 都經過了小分子或蛋白質的資料集訓練,但這個專案是首批以 DNA 和 RNA 的最小單位核苷酸序列訓練的模型之一。
該專案的負責人、來自阿貢國家實驗室的運算生物學家 Arvind Ramanathan 表示:「我們假設從蛋白質層級到基因層級的資料可能有助於我們建立更完善的模型來瞭解 COVID 變異株。我們訓練模型追蹤整個基因體及其進化過程中出現的所有變化,可以因此做出更準確的預測,不僅是 COVID,任何具有足夠基因組資料的疾病都是如此。」
戈登貝爾獎被視為高效能運算領域的諾貝爾獎,電腦協會 (Association for Computing Machinery) 在其舉辦的 SC22 大會上舉行該頒獎儀式,該協會代表著全球約 100,000 名運算專家。自 2020 年以來,該協會即針對用運高效能運算提升對 COVID 理解的傑出研究頒發特別獎。
以四個字母的語言訓練 LLM
LLM 長久以來都是以人類語言訓練,通常由幾十個字母組成,可以排列成數萬個詞彙,並結合成更長的句子和段落。另一方面,生物學的語言中,只有四個代表核苷酸的字母,包括 DNA 中的 A、T、G 和 C,或 RNA 中的 A、U、G 和 C,由這些字母排列成不同的基因序列。
雖然字母較少,對人工智慧來說看似比較簡單,但生物學的語言模型其實要複雜得多,因為人類體內由超過 30 億個核苷酸組成基因組,以及冠狀病毒中約 30,000 個核苷酸組成基因組,很難分解成不重複且有意義的單位。
Ramanathan 表示:「在瞭解生命的奧秘時會面臨一項重大挑戰,那就是基因組中的定序資訊相當龐大。核苷酸序列的含義可能會受到另一個序列的影響,這比人類書寫文字中所使用的句子或段落間的差異更大,甚至可以相當於書中章節的關係。」
參與這個專案的 NVIDIA 協作者設計了一種分層漫射法,使 LLM 能夠將大約由 1,500 個核苷酸組成的長字符串當作句子處理。
論文的共同作者、NVIDIA 人工智慧研究資深總監、加州理工學院運算與數學科學系及加州大學爾灣分校 Bren 學院教授 Anima Anandkumar 表示:「標準語言模型很難產生一致的長序列,並學習不同變異的基本分布。我們開發出能以更高細節程度運作的漫射模型,因此可產生逼真變異,並擷取更佳的統計資料。」
預測令人擔憂的 COVID 變異株
團隊使用來自細菌和病毒生物資訊中心的開放原始碼資料,首先是預先訓練了超過 1.1 億個基因序列的 LLM,這些基因序列皆來自像是細菌這種單細胞生物的原核生物,接著再使用 150 萬個 COVID 病毒的高品質基因體序列微調模型。
研究人員在更廣泛的資料集上預先訓練,藉此確保他們的模型可通則化並應用於未來其他專案的預測工作,使其成為首批具備此功能的全基因組規模模型之一。
經過 COVID 資料微調後,LLM 便能夠分辨病毒變異株的基因體序列,也能夠產生自己的核苷酸序列,預測 COVID 基因組的潛在變異,有助於科學家預測未來令人擔心的變異株。
Ramanathan 表示:「大多數的研究人員都一直在追蹤 COVID 病毒的棘蛋白變異,尤其是與人體細胞結合的領域。但是病毒基因組中還有其他蛋白質會經歷頻繁的變異,理解這些變異也很重要。」
論文指出,該模型也可以與 AlphaFold 和 OpenFold 等熱門的蛋白質結構預測模型整合,協助研究人員模擬病毒結構,並研究基因變異如何影響病毒感染宿主的能力。OpenFold 是 NVIDIA BioNeMo LLM 服務中包含的預先訓練語言模型之一,適用於將 LLM 應用於數位生物學和化學應用程式的開發人員。
利用 GPU 加速的超級電腦大幅強化人工智慧訓練
團隊在採用 NVIDIA A100 Tensor 核心 GPU 的超級電腦上開發人工智慧模型,包括阿貢國家實驗室的 Polaris、美國能源部的 Perlmutter 以及 NVIDIA 的內部 Selene 系統。這些超級電腦擴充了這些強大的系統,在執行訓練時能達到超過 1,500 Exaflops 的效能,創造至今規模最大的生物語言模型。
Ramanathan 表示:「我們今天使用的這些模型有多達 250 億個參數,我們預計未來這個數量會顯著增加。從模型大小、基因序列長度和需要的訓練資料量,就可以得知我們確實需要具備數千個 GPU 的超級電腦提供的計算複雜性。」
研究人員估計,在大約 4,000 個 GPU 上,使用 25 億個參數訓練模型花了一個多月的時間。這個團隊已經在研究生物學 LLM,並在公開發表論文和程式碼之前,花了大約四個月的時間完成專案。GitHub 頁面上有其他研究人員在 Polaris 和 Perlmutter 上執行模型的說明。
NVIDIA BioNeMo 框架透過 NVIDIA NGC 中樞提供搶先體驗版,支援針對 GPU 最佳化的軟體,協助研究人員在多個 GPU 上擴充大型生物分子語言模型。該框架作為 NVIDIA Clara Discovery 藥物研發工具集的一部分,將支援化學、蛋白質、DNA 和 RNA 資料格式。
與 NVIDIA 一起參與 SC22 大會,並在下方觀看特別演講的重播:
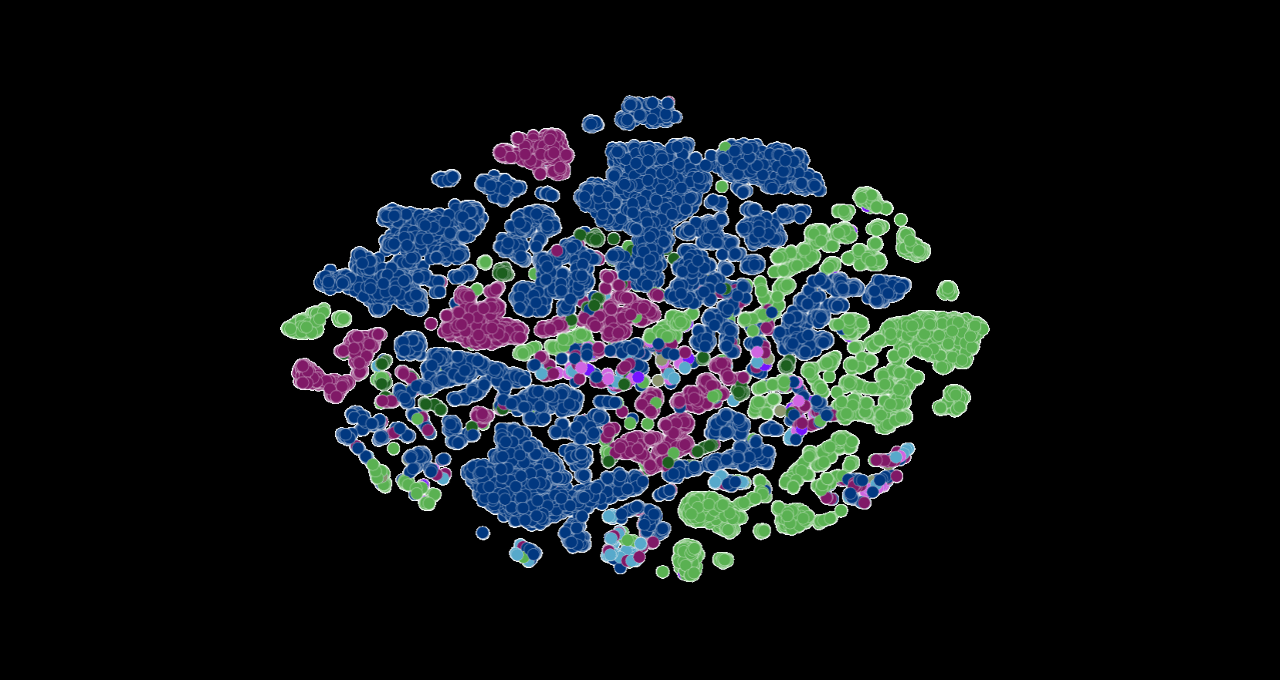
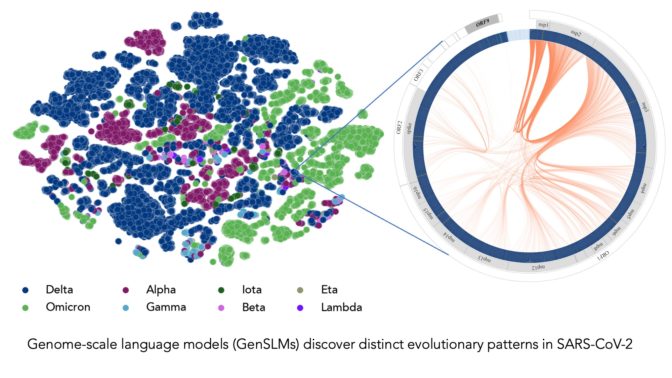
上方的影像代表由研究人員 LLM 排序的 COVID 病毒株。每個點都按 COVID 變異株以顏色編碼。影像來源:阿貢國家實驗室的 Bharat Kale、Max Zvyagin 和 Michael E. Papka。